载脂蛋白E(ApoE)与阿尔兹海默病的调节
载脂的ApoE脂蛋白颗粒与多种细胞表面受体结合,支持脑膜稳态和损伤修复。从患病率和相对危险程度来看,ApoE基因的ε4等位基因是迟发性阿尔茨海默病(AD)最为突出的遗传风险因素。ApoE4通过多种途径参与AD的发病,这些途径包括但不限于淀粉样蛋白-β(Aβ)肽的代谢、聚集和毒性,以及tau病、突触可塑性、脂质转运、葡萄糖代谢、线粒体功能、血管完整性和神经炎症。在探索阿尔茨海默病的病理生理学时,对ApoE相关途径的新认识为开发有效的AD治疗手段开辟了新的途径与可能性。
1.阿尔茨海默病与ApoE
阿尔茨海默病(AD)作为痴呆症最常见的类型,是一种神经退行性疾病,其标志性特征包括细胞外老年斑的形成以及细胞内神经原纤维缠结的堆积,同时伴随着记忆力、思维能力、语言功能的逐步衰退及行为模式的改变。日益增多的遗传学与生物化学证据表明,淀粉样蛋白β(Aβ,具体由40或42个氨基酸序列构成)在大脑中的逐渐累积与集结,是淀粉样蛋白前体蛋白(APP)经过特定蛋白水解途径分解后的直接产物,这一机制在AD的发病过程中起到了至关重要的作用。毒性可溶性Aβ寡聚体、神经元内部蓄积的Aβ以及细胞外形成的淀粉样蛋白斑块,均会严重干扰正常的突触功能,并激活一系列下游毒性信号通路,最终促使神经细胞发生变性。值得注意的是,Aβ诱导的神经元功能障碍的毒性效应,可能还与微管相关蛋白tau的共存状态密切相关,该蛋白在AD患者的大脑中以神经原纤维缠结的形式异常聚集,进一步加剧了疾病的进展。
仅有极少数(不足5%)的阿尔茨海默病(AD)病例会在65岁之前显现出症状,这类情况通常被称作早发性AD(EOAD)。EOAD中有一类尤为引人注目的类型,即家族性AD(FAD),它是由APP、早老素1(PSEN1)或PSEN2基因中的特定基因突变所触发的,这些突变会直接导致β淀粉样蛋白(Aβ)的异常过量产生。相比之下,绝大多数的AD病例均发生在65岁及以上的老年人群中,被称为晚发型AD(LOAD)。在LOAD的发病机制中,Aβ的清除障碍似乎成为了Aβ在脑内积聚的首要原因。
从患病率和相对风险性的角度来看,LOAD最为显著的遗传风险因素当属载脂蛋白E(ApoE)基因的等位基因变异。具体而言,与常见的ε3等位基因相比,携带ε4等位基因的人群罹患AD的风险会显著增加,而拥有ε2等位基因则似乎具有一定的保护作用。ApoE4可能通过多种不同的机制发挥其毒性作用,从而进一步提高AD的发病风险。
2.ApoE的生化特征
ApoE是一种由299个氨基酸构成的糖蛋白,分子量大约为34kDa。它通过与细胞表面的ApoE受体相结合,在血浆及中枢神经系统(CNS)中有效地运输并递送胆固醇及其他脂质物质。ApoE的受体结合区域定位于N端结构域(涵盖残基136-150),而其主要的脂质结合区域(涵盖残基244-272)则位于C端结构域。值得注意的是,ApoE存在三种同工异构体:ApoE2、ApoE3和ApoE4,它们在112号和158号位置的氨基酸残基上存在差异。具体而言,ApoE2在这两个位置上都拥有Cys残基;ApoE3在112位为Cys残基,而在158位则是Arg残基;ApoE4则在两个位置上都为Arg残基。
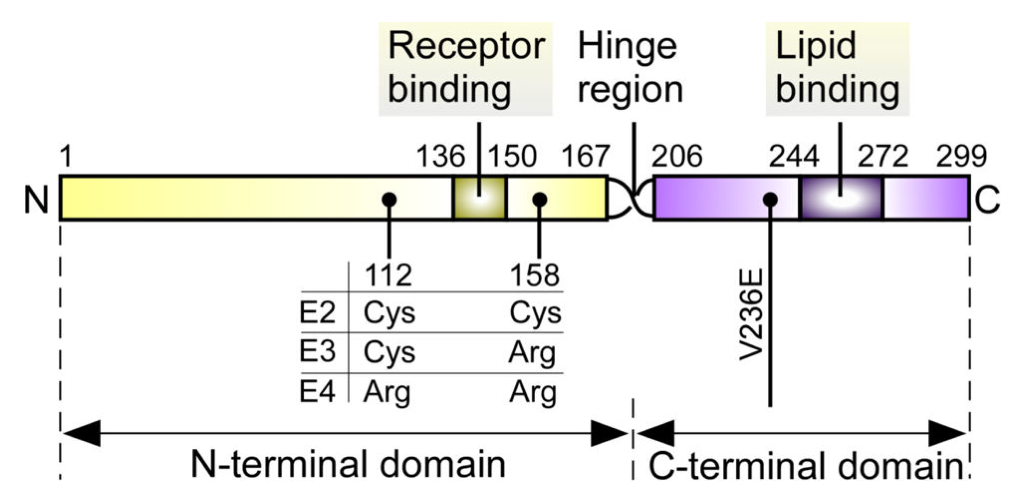
图1.ApoE结构示意图
注:脂质结合区外的V236E变异可降低晚发性阿尔茨海默病(LOAD)风险。
ApoE主要由外周组织中的肝细胞、巨噬细胞以及脂肪细胞合成产生。在CNS中,ApoE的表达通常受到严格调控,在应激或损伤等特定条件下,会在星形胶质细胞、小胶质细胞、血管壁细胞以及脉络丛细胞中高度表达。分泌后的ApoE会被ATP结合盒转运蛋白A1(ABCA1)和ABCG1进行脂质化修饰,这些转运蛋白将细胞内的胆固醇和磷脂转移至新生的ApoE上,从而形成脂蛋白颗粒。在血浆中,ApoE更倾向于与极低密度脂蛋白(VLDL)颗粒结合,而在中枢神经系统的HDL样颗粒中也检测到了ApoE的存在。脑内的ApoE主要来源于局部的合成,因为血脑屏障(BBB)对ApoE的跨膜转运具有严格的限制作用。
不同ApoE基因型个体的血浆ApoE水平存在差异,其中ApoE ε2/2基因型的水平最高,而ApoE ε4/4基因型的水平最低。这种亚型依赖的差异同样在脑脊液和间质液(ISF)中得到了体现。此外,通过精氨酸-61和谷氨酸-255之间的离子相互作用,ApoE4的结构域间相互作用可能会增强其肝脏清除效率,并促进星形胶质细胞对其的降解过程。
3.ApoE与AD Aβ病理学
3.1ApoE对脑Aβ沉积的影响
临床研究报告指出,在认知功能正常的个体、轻度认知障碍(MCI)患者以及已出现症状的阿尔茨海默病(AD)患者中,大脑内Aβ沉积的程度与ApoE4基因型的存在呈正相关关系。值得注意的是,在认知功能尚未受损的ApoE4携带者中(平均年龄约为56岁),淀粉样蛋白成像结果显示其Aβ沉积的出现时间早于ApoE4非携带者(平均年龄为76岁)。这些发现进一步提示,ApoE4基因可能通过促使Aβ在大脑中的更早积累、加速其聚集及沉积过程,从而增加个体罹患阿尔茨海默病的风险。
3.2ApoE对CSF Aβ的影响
脑脊液(CSF)中的Aβ作为一种生物标志物,展现出了极高的潜力,其准确性足以有效区分阿尔茨海默病(AD)患者与对照组或罹患其他神经系统疾病的患者。尽管已知ApoE4等位基因与大脑中Aβ沉积的增加有关,但关于它如何具体影响CSF中Aβ水平的机制,目前仍存在争议。早期的研究揭示,ApoE4与AD患者及对照组中CSF Aβ42水平的降低之间存在着紧密的关联。然而,近期一项研究发现:CSF Aβ42水平与AD的诊断及脑内Aβ的积累密切相关,而这一关系似乎并不受ApoE基因型的影响。此外,还有研究聚焦于CSF中ApoE与Aβ42水平的关系,发现ApoE蛋白水平与CSF中的Aβ42以及磷酸化tau水平均呈正相关,这进一步提示ApoE或可成为AD的另一种潜在生物标志物。
4.ApoE调节Aβ代谢的机制
大脑中Aβ的水平体现了Aβ生成与清除之间的净平衡状态;阿尔茨海默病(AD)患者大脑中Aβ的累积,可能是由于Aβ的过度生成、清除效率低下,或是这两种情况同时存在所导致的。越来越多的证据表明,ApoE及其受体在这些调节过程中起重要作用。
4.1ApoE与Aβ产生
APP定位于质膜表面,在此处它经历α-分泌酶的蛋白水解作用。在淀粉样蛋白的生成路径中,APP首先被内吞,随后由β-分泌酶和γ-分泌酶裂解,最终产生Aβ肽(Aβ40和Aβ42)。ApoE受体(包括LRP1)与APP相互作用,并调控其转运过程以及向Aβ的转化。ApoE以亚型特异的方式调节APP的运输及Aβ的产生。在神经元细胞系中,相较于ApoE3,ApoE4更能促进APP的内化作用,从而进一步增加Aβ的生成量。据报道,ApoE同工型(ApoE4>ApoE3>ApoE2)通过激活人神经元中的非经典丝裂原活化蛋白激酶(MAPK)信号转导途径,对APP的转录及Aβ的生成产生不同程度的影响。然而,这些影响是否最终会增加罹患阿尔茨海默病(AD)的风险,仍需进一步的深入研究。
4.2ApoE与Aβ清除
Aβ主要由神经元产生。可溶性Aβ在脑实质中的积累导致Aβ寡聚体和淀粉样蛋白斑块的形成,而它在血管周围区域的积累会引发CAA。为了从大脑中清除这些可溶性Aβ,存在多种紧密相关的清除机制:ISF中的可溶性Aβ可以在脑实质中清除,通过BBB转运至血液,或由血管壁细胞降解。此外,Aβ还可以沿着ISF流进入CSF,最终被循环和淋巴系统吸收。Aβ的主要清除通路,包括脑实质细胞(神经元和神经胶质细胞)通过受体(如LRP1和LDLR)介导清除,以及血管壁细胞从血管周围间隙摄取,还有内肽酶(例如NEP、IDE)蛋白水解降解。
4.2.1ApoE亚型对受体介导的Aβ清除的影响
ApoE主要由神经胶质细胞产生,并通过ABCA1和ABCG1转运蛋白脂质化,形成脂蛋白颗粒 。这些脂化的ApoE能够与Aβ结合,并通过细胞表面受体(例如LRP1、LDLR)被内化,促进Aβ清除。 大多数内吞的ApoE被回收,而Aβ通常被降解。由于ApoE4-Aβ复合物的稳定性低于ApoE3-Aβ,因此ApoE4在促进Aβ清除方面不如ApoE3有效(ApoE2>ApoE3>ApoE4)。此外,ApoE也可能通过与Aβ竞争细胞表面受体来抑制Aβ清除,ApoE4相对于其他ApoE同工型表现出最强的清除阻断作用(ApoE4>ApoE3)。
4.2.2ApoE亚型对蛋白酶介导的Aβ清除的影响
Aβ可通过蛋白水解酶降解从大脑中去除,这些蛋白酶包括脑啡肽酶(NEP)、胰岛素降解酶(IDE)、内皮素转换酶(ECE)、血管紧张素转换酶(ACE)、纤溶酶原激活剂、基质金属蛋白酶2(MMP2)以及MMP9 。ApoE可促进大脑中可溶性Aβ的蛋白水解清除,其中ApoE3促进作用更强。值得注意的是,当Aβ与细胞表面HSPG结合时,会阻碍Aβ的蛋白水解降解,促进Aβ寡聚化和聚集 。
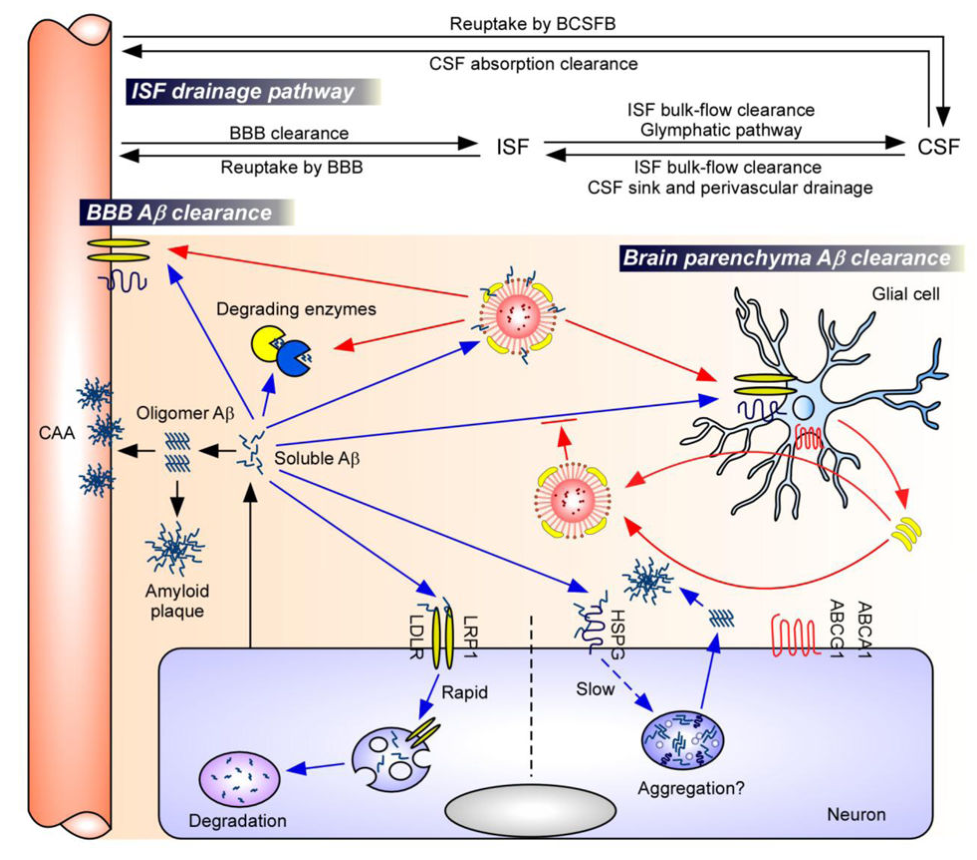
图2.主要的Aβ清除途径以及ApoE和ApoE受体的作用
缩写:Aβ,淀粉样蛋白-β;BBB,血脑屏障;BCSFB,血液-CSF屏障;CSF,脑脊液;ISF,间质液;CAA,脑淀粉样血管病;NEP,脑啡肽酶;IDE,胰岛素降解酶;LRP1,低密度脂蛋白受体相关蛋白1;LDLR,低密度脂蛋白受体;HSPG,硫酸乙酰肝素蛋白多糖;ABCA1,ATP结合盒A1;ABCG1,ATP结合盒G1。
参考文献
Zhao N, Liu CC, Qiao W, et al. Apolipoprotein E, Receptors, and Modulation of Alzheimer's Disease[J]. Biol Psychiatry. 2018,83(4):347-357.

相关产品

 苏公网安备32011202001302
苏公网安备32011202001302